A new, lightning fast implementation of ADMIXTOOLS.
Overview
ADMIXTOOLS is a collection of programs which use genetic data to infer how populations are related to one another. It has been used in countless publications to test whether populations form clades (qpDstat, qpWave), to estimate ancestry proportions (qpAdm), and to fit admixture graphs (qpGraph).
ADMIXTOOLS 2 provides the same functionality as ADMIXTOOLS in a new look, and it’s orders of magnitude faster. This is achieved through separating the computation of f2-statistics from all other computations, and through a number of other optimizations.
Features
- Much faster than the original ADMIXTOOLS software
- Simple R command line interface
- Even simpler point-and-click interface
- Several new features and methodological innovations that make it easier to find robust models:
- Automated and semi-automated admixture graph inference
- Simultaneous exploration of many qpAdm models
- Unbiased comparison of any two qpGraph models using out-of-sample scores
- Jackknife and bootstrap standard errors and confidence intervals for any qpAdm, qpWave, and qpGraph parameters
- Interface with msprime makes it easy to simulate genetic data under an admixture graph
- Full support for genotype data in PACKEDANCESTRYMAP/EIGENSTRAT format and PLINK format
- Wrapper functions around the original ADMIXTOOLS software (see also admixr)
- Extensive documentation
- New features available on request!
Installation
install.packages("devtools") # if "devtools" is not installed already
devtools::install_github("uqrmaie1/admixtools")
library("admixtools")The above commands will install all R package dependencies which are required to run ADMIXTOOLS 2 on the command line. For the interactive app, additional packages are required, which can be installed like this:
devtools::install_github("uqrmaie1/admixtools", dependencies = TRUE)If you encounter any problems during the installation, this is most likely because some of the required R packages cannot be installed. If that happens, try manually re-installing some of the larger packages, and pay attention to any error message:
install.packages("Rcpp")
install.packages("tidyverse")
install.packages("igraph")
install.packages("plotly")Running devtools::install_github("uqrmaie1/admixtools") will compile C++ from source code (this isn’t necessary when installing packages from CRAN with install.packages()).
If you get the following error:
Error: Failed to install 'admixtools' from GitHub: Could not find tools necessary to compile a package.
you might be able to solve the problem by installing Rtools for your version of R if you use Windows, or Xcode command line tools if you use macOS. This is also described here.
On some Linux distributions, it may be necessary to manually install additional libraries or programs. For example, if you see the error gfortran: command not found during installation, you would have to install gfortan with sudo zypper in gfortran or sudo apt-get install gfortran, depending on the Linux distribution.
If you get any other errors during installation, please contact me.
Usage
Admixture graphs can be fitted like this:
genotype_data = "/my/geno/prefix"
fit = qpgraph(genotype_data, example_graph)
fit$score
plot_graph(fit$edges)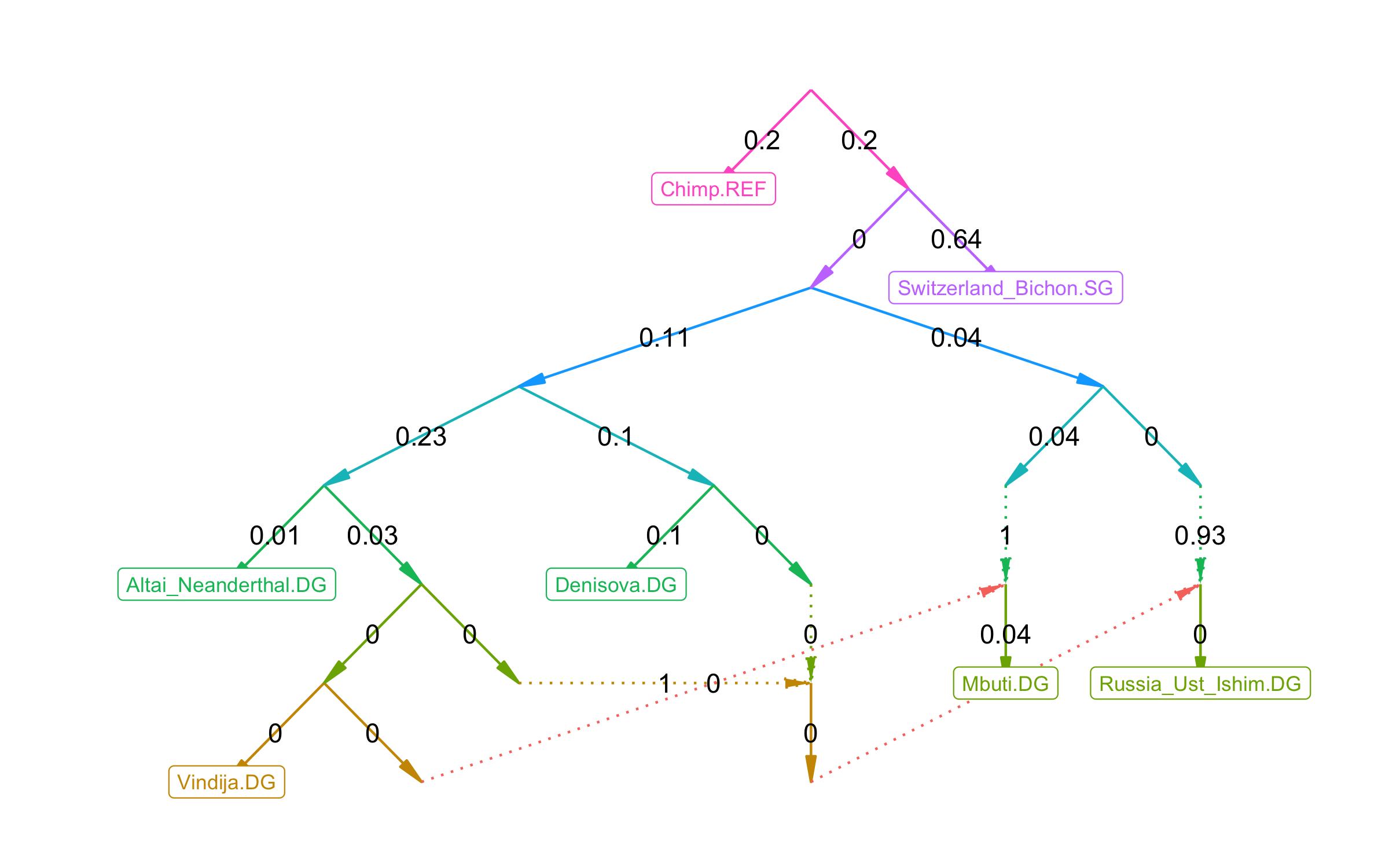 Clearly not a historically accurate model, but it gets the idea across.
Clearly not a historically accurate model, but it gets the idea across.
When testing more than one model, it makes sense to extract and re-use f2-statistics:
f2_blocks = f2_from_geno(genotype_data)
fit = qpgraph(f2_blocks, example_graph)
fit$score
#> [1] 19219.98f2-statistics can also be used to estimate admixture weights:
left = c("Altai_Neanderthal.DG", "Vindija.DG")
right = c("Chimp.REF", "Mbuti.DG", "Russia_Ust_Ishim.DG", "Switzerland_Bichon.SG")
target = "Denisova.DG"
qpadm(f2_blocks, left, right, target)$weights#> # A tibble: 2 × 5
#> target left weight se z
#> <chr> <chr> <dbl> <dbl> <dbl>
#> 1 Denisova.DG Altai_Neanderthal.DG 49.6 23.3 2.13
#> 2 Denisova.DG Vindija.DG -48.6 23.3 -2.08Or to get f4-statistics:
f4(f2_blocks)#> # A tibble: 105 × 8
#> pop1 pop2 pop3 pop4 est se z p
#> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl>
#> 1 Altai_Neanderthal.DG Chimp.REF Deni… Mbut… 0.0196 6.07e-4 32.4 1.32e-229
#> 2 Altai_Neanderthal.DG Denisova.DG Chim… Mbut… -0.0129 3.64e-4 -35.6 2.22e-277
#> 3 Altai_Neanderthal.DG Mbuti.DG Chim… Deni… -0.0326 5.22e-4 -62.5 0
#> 4 Altai_Neanderthal.DG Chimp.REF Deni… Russ… 0.0180 6.87e-4 26.3 6.43e-152
#> 5 Altai_Neanderthal.DG Denisova.DG Chim… Russ… -0.0152 4.46e-4 -34.0 4.67e-254
#> 6 Altai_Neanderthal.DG Russia_Ust… Chim… Deni… -0.0332 5.55e-4 -60.0 0
#> 7 Altai_Neanderthal.DG Chimp.REF Deni… Swit… 0.0181 6.63e-4 27.3 1.09e-164
#> 8 Altai_Neanderthal.DG Denisova.DG Chim… Swit… -0.0150 4.64e-4 -32.3 6.06e-229
#> 9 Altai_Neanderthal.DG Switzerlan… Chim… Deni… -0.0331 5.74e-4 -57.7 0
#> 10 Altai_Neanderthal.DG Chimp.REF Deni… Vind… -0.0771 6.98e-4 -110. 0
#> # ℹ 95 more rowsInteractive browser app
ADMIXTOOLS 2 also has a simple point-and-click interface. This makes it easy to explore many qpAdm or qpGraph models at the same time, for example by allowing you to build and change admixture graphs interactively. Typing the following command in the R console launches the app:
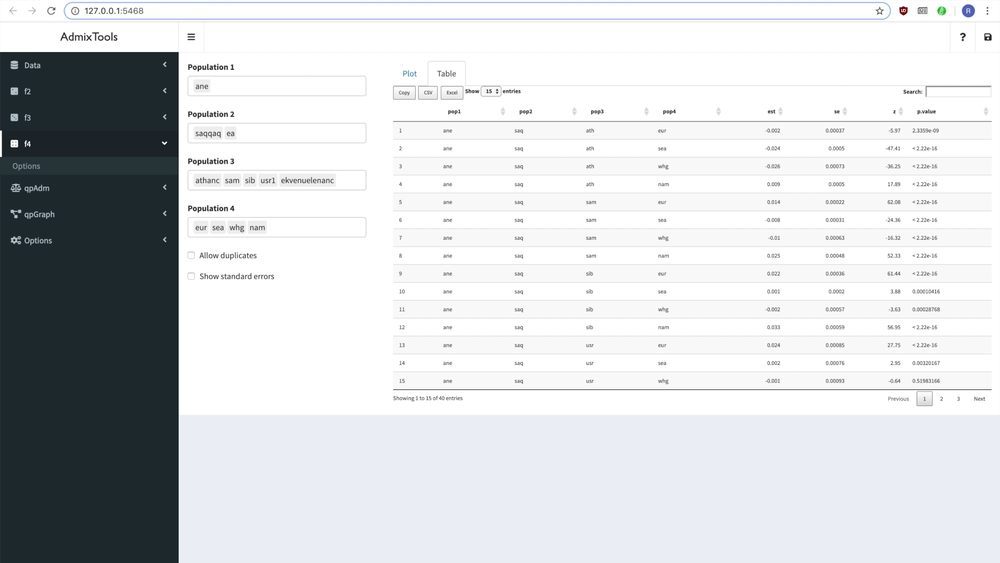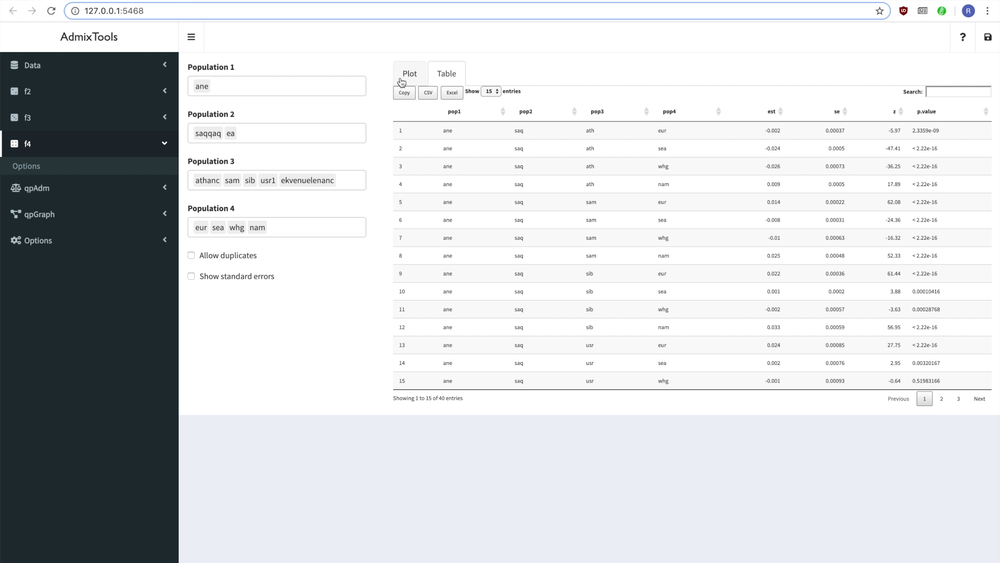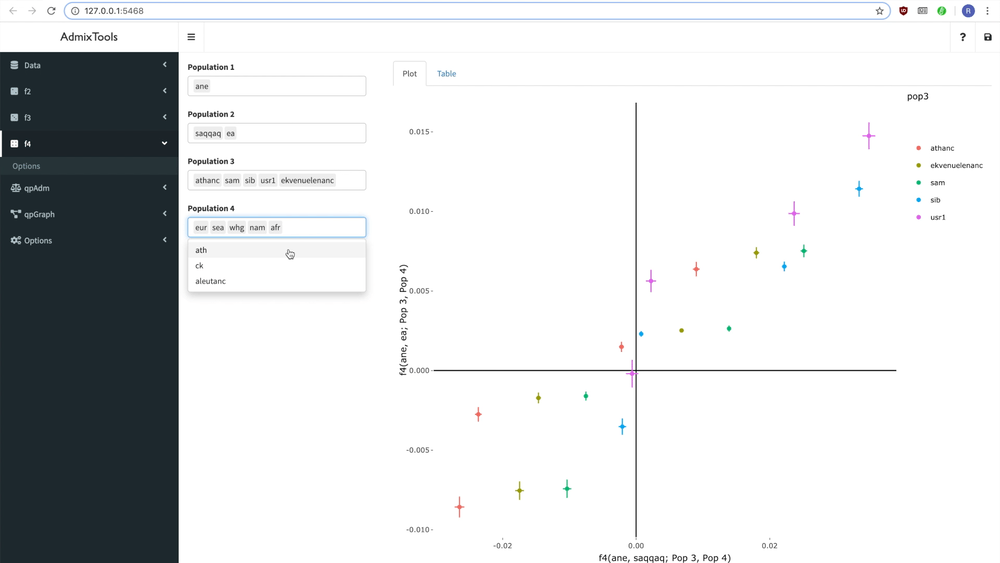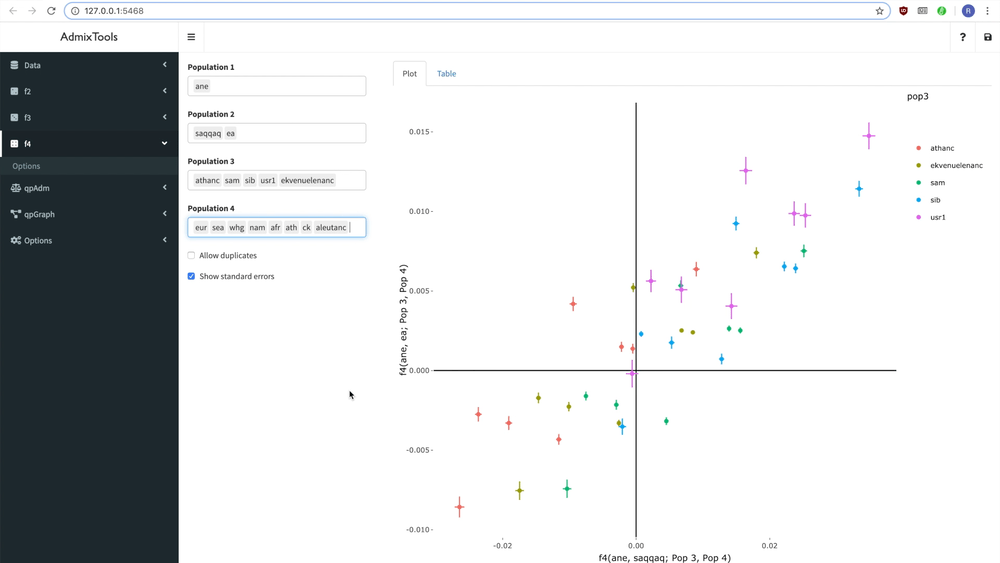
Documentation
One of the design goals behind ADMIXTOOLS 2 is to make the algorithms more transparent, so that the steps leading from from genotype data to conclusions about demographic history are easier to follow.
To this end, all ADMIXTOOLS 2 functions are documented. You can also take a look at the tutorial, read more about how ADMIXTOOLS 2 computes f-statistics and standard errors, and what you can do with admixture graphs.
For even greater transparency, many of the core functions are implemented twice: In C++ for performance (used by default), and in R, which makes it easier to trace the computations step by step. For example, if you want to know how weights are computed in qpadm(), you can type qpadm in R to get the function code and you will see another function which is called qpadm_weights(). By default, this function will be replaced by its faster C++ version cpp_qpadm_weights(). But you can still see what it’s doing without reading the C++ code by typing admixtools:::qpadm_weights. And you can tell qpadm() to use the R versions instead of the C++ versions by calling qpadm(cpp = FALSE).
Contact
For questions, feature requests, and bug reports, please submit an issue on GitHub, or contact Robert Maier or Pavel Flegontov.
Acknowledgments
Nick Patterson has developed the original ADMIXTOOLS software. David Reich and Nick Patterson have not only conceived most of the ideas that have made ADMIXTOOLS so successful, they have also provided me (and continue to provide me) with critical ideas and feedback in the development of ADMIXTOOLS 2.
I also want to thank all members of the Reich lab for many discussions which have helped me gain a better understanding of the underlying methods, and for crucial feedback about ADMIXTOOLS 2; in particular Pavel Flegontov, Iosif Lazaridis, Mark Lipson, Harald Ringbauer, Shop Mallick, and Tian Chen Zeng.
Many useful features in ADMIXTOOLS 2 were suggested by its users. If you are able to install and run ADMIXTOOLS 2 without issues, it is only because others have cleared the path for you, by bringing problems to my attention. In no particular order, I want to thank Ornob Alam, Ming-Shan Wang, Angad Johar, Ezgi Altınışık, Tobias Göllner, Christian Huber, Kale, Ted Kandell, Fraser Combe, Matthew Williams, Lenny Dykstra, Kristján Moore, Dilawer Khan, Lareb Humayoun, Sánta Benedek, Steven Rosenberg, Daniel Tabin, Benjamin Peter, and Ahmad Bekhit.
If you expected to see your name here but I failed to include it, please let me know about it!
Cite ADMIXTOOLS 2
For referencing ADMIXTOOLS 2 in your work, please cite our eLife paper.
There is an earlier preprint of the manuscript, and some additional personal comments here.
See also
- ADMIXTOOLS The original ADMIXTOOLS software
- admixr An R package with ADMIXTOOLS wrapper functions and many useful tutorials
- admixturegraph An R package for automatic graph inference
- miqoGraph A Julia package for automatic graph inference
- MixMapper Another method to infer admixture graphs
- TreeMix Another method to infer admixture graphs
- Legofit A program to estimate the history of population size, subdivision, and gene flow
- qpBrute Automated graph fitting and Bayes factor calculations